
Translate this page into:
A comprehensive compatibility study of ganciclovir with some common excipients

*Corresponding author: Ashok Kumar Yadav, University Institute of Pharmaceutical Sciences, Panjab University, Chandigarh, India. ashoky@pu.ac.in
-
Received: ,
Accepted: ,
How to cite this article: Mishra A, Sinha VR, Sharma S, Mathew AT, Kumar R, Yadav AK. A comprehensive compatibility study of ganciclovir with some common excipients. Am J Biopharm Pharm Sci. 2023;3:2. doi: 10.25259/AJBPS_4_2023
Abstract
Objectives:
The aim of the present study is to illustrate compatibility testing of ganciclovir (GCV) with some common excipients that would be used to manufacture solid oral dosage forms. Different spectroscopy techniques were utilized to see the interaction of GCV with excipients such as lactose, microcrystalline cellulose (MCC), magnesium stearate, and talc, and dicalcium phosphate. Further, a molecular docking study was also done to know the interaction of GCV with excipients. In vitro study of a physical mixture of GCV with excipients was performed to get the release of drug.
Material and Methods:
A number of analytical techniques (differential scanning calorimetry [DSC] using DSC-Q20, TA instruments, Fourier-transform infrared spectroscopy [FTIR] spectroscopy using Spectrum RX 1, nuclear magnetic resonance [NMR] using Bruker Advance Neo 500 MHz NMR spectrometer, etc.) have been used to explore the drug-excipient compatibility. Further, a suspected interaction was evaluated by thin-layer chromatography (TLC). In vitro dissolution studies in different sets of experiments were accomplished to determine the influence of hydrophobic and hydrophilic attributes of excipients (MCC, lactose, dicalcium phosphate, and talc) on the dissolution profile of GCV using USP1-type dissolution apparatus. Furthermore, in silico molecular docking studies were also performed to evaluate any probable molecular interactions among drugs and excipients using Auto Dock VINA 1.2.0 software and GROMACS 5.0 software.
Results:
Comparing FTIR and 1H NMR spectra of GCV and physical mixtures of GCV and excipients, no significant deviation of characteristic peaks in infrared spectroscopy and 1H NMR signals was observed. The DSC of GCV showed two sharp endothermic peaks at 238.82°C and 255°C. The endothermic peak of GCV in DSC thermogram of physical mixtures was observed in nearly the same position except with lactose and dicalcium phosphate. A slightly deviated peak of GCV with a physical mixture of drug and lactose and dicalcium phosphate indicated that there were suspected interactions between the drug with lactose and dicalcium phosphate. These interactions were evaluated by thin-layer chromatography (TLC) and it confirmed that there was no interaction between drugs and excipients. In vitro dissolution studies determined the influence of hydrophobic and hydrophilic attributes of excipients on the dissolution profile of GCV. The physical mixture of GCV with MCC displayed a maximum amount (66.48%) of drug release in 10 min. On the other hand, a physical mixture of GCV with talc showed a minimum amount (12.08%) of drug release in 10 min. Docking study predicted that the number of interactions were more between GCV and lactose (four nos.) in comparison to GCV and MCC (two nos.). This interaction supported the in vitro drug release of a physical mixture of GCV with MCC which was higher than a mixture of GCV with lactose.
Conclusion:
Compatibility testing of GCV with used excipients by analytical techniques confirmed that GCV should be compatible with used excipients. Drug dissolution of GCV and physical mixture of MCC exhibited the maximum amount of drug release whereas a mixture of GCV with talc released the minimum amount of drug for both short (10 min.) and long (60 min.) periods. Docking studies disclosed that the lactose complex showed less deviation with less root mean square deviation value in comparison to the microcrystalline complex. Thus, the lactose complex has more hydrogen bonds and it was more stable as compared with the MCC complex. GCV indicates that the total energy of the MCC complex is less than that of the lactose complex. This indicates that GCV is more soluble when combined with the microcrystalline complex. Therefore, GCV and used excipients could be used for solid dosage formulations.
Keywords
Ganciclovir
Compatibility studies
Molecular docking
Excipients
In vitro dissolution

INTRODUCTION
The quality, safety, efficacy, and stability of any pharmaceutical product are paramount in any new drug development process. To ensure consistency in these parameters, comprehensive information about active pharmaceutical ingredients and their excipients is necessary.[1] An excipient is considered as the vital component of a quality drug product.[2] Any form of physicochemical interaction between the drug and excipient will potentially affect the product stability and bioavailability.[3] Therefore, drug-excipient compatibility study is a prerequisite for the development of drugs that are safe and stable for use. Proper selection and assessment of possible incompatibilities among the drug and excipients during preformulation studies are of paramount importance to accomplish the target product profile and critical quality attributes.[4,5] To avoid incompatibilities, problems encountered during drug development and post-commercialization, there is a need for proper assessment of possible incompatibilities among the drug and excipients using appropriate analytical technique.[6] These analytical techniques are needed not only to generate useful information with regard to which excipient is compatible with a drug substance but also for troubleshooting unexpected problems which might arise during formulation processes.[7,8]
The present study aims to evaluate the drug-excipient compatibility testing of ganciclovir (GCV) using modern analytical techniques. GCV (an antiviral drug) was selected for studying the effect of drug-excipient compatibility study. Ganciclovir (GCV) is 26 times more potent than its analog acyclovir against the human cytomegalovirus (CMV) strain in in vitro. The drug has antiviral properties which are primarily used for herpes simplex virus and CMV and are also used to treat different types of other viruses such as varicella-zoster virus and Epstein-Barr virus.[9] The excipients, which are used in the present study, are lactose (LAC), magnesium stearate, microcrystalline cellulose (MCC), talc, and dicalcium phosphate. The interaction between the drug-excipient was evaluated by physicochemical studies that would be done using Fourier-transform infrared spectroscopy (FTIR), nuclear magnetic resonance (NMR), and differential scanning calorimetry (DSC). The dissolution behavior of the drug (GCV) was also studied to find out the amount of drug release with different excipients in each physical mixture. Further, a thin layer chromatography (TLC) study was done to confirm the possible interaction between the drug and excipient. To support compatibility testing and drug release in vitro, a docking study was also carried out to find the possible interaction between excipients and drugs. GCV compatibility studies were conducted with lactose, MCC, talc, dicalcium phosphate, mannitol, and magnesium stearate and this compatibility study was not reported for GCV with the above-mentioned excipients.
MATERIAL AND METHODS
Materials
GCV was obtained from Ranbaxy Pvt. Ltd., New Delhi, India, as a gift sample for research purposes; and lactose, MCC, magnesium stearate, talc, and dicalcium phosphate were purchased from Loba Chemie Pvt. Ltd., Mumbai, and Central drug house, New Delhi, respectively. The solvents were obtained from Thermo-Fisher Scientific India Pvt. Ltd., Mumbai, India.
Methods
Compatibility studies
Compatibility studies of GCV with common excipients were done by preparing a physical mixture of GCV with excipients in the ratio of 1:1 and examined using the following analytical techniques.
FTIR studies
FTIR spectroscopy identifies the characteristic vibrational frequencies exhibited by various functional groups present in the test compound. FTIR can be useful for the evaluation of compatibility between components since any modification in the bond that exhibits characteristic vibrational frequencies produces spectral changes leading to frequency shifts and splitting in the absorption peaks. Thus, the presence of any spectral changes during the compatibility study might be an indication of probable incompatibility. KBr disk method (sample to KBr weight ratio of 1:100) was used to obtain the FTIR spectra using Spectrum RX 1, (Perkin Elmer, U.K.) in a range of 4000 cm−1–500 cm−1.[10]
DSC studies
DSC thermograms of samples were obtained using a DSC-Q20, TA instruments (New Castle Delaware, U.S). The method was followed as reported in the literature with certain modifications. Samples were heated in the sealed standard aluminum pans from 50°C to 300°C at a scanning rate of 10°C/min under nitrogen purge, with an empty aluminum pan as reference. DSC thermograms displayed interactions if there were appearance or disappearance of new endothermic peaks, and significant shifts in the melting point of components or variations in corresponding enthalpies of reaction were observed.[11,12]
NMR spectroscopy studies
NMR is the most widely used technique to investigate the structure of molecules. NMR is quite a sensitive technique to know the molecular structural features as the neighboring atoms influence the signals from individual nuclei. The NMR spectra of GCV and drug-excipient physical mixtures (1:1) were recorded in DMSO-d6 using Bruker Advance Neo 500 MHz NMR spectrometer.[13-15]
TLC studies
TLC is a method for analyzing mixtures by separating the compounds in the mixture. TLC is a sensitive technique and a microgram (0.000001 g) quantity can be analyzed by TLC and it takes little time for an analysis (about 5–10 min). TLC consists of three steps-spotting, development, and visualization. First, the sample to be analyzed is dissolved in a volatile (easily evaporated) solvent to produce a very dilute (about 1%) solution. Spotting consists of using a micropipette to transfer a small amount of this dilute solution to one end of a TLC plate, and in this case, a thin layer of powdered silica gel that has been coated onto a plastic sheet. The spotting solvent quickly evaporates and leaves behind a small spot of the material. The development consists of placing the bottom of the TLC plate into a shallow pool of development solvent, which, then, travels up the plate by capillary action. The mobile phase that was used in this study was methanol: CHCl3 in the ratio of 2:3. As the solvent travels up to the plate, it moves over the original spot. A competition is set up between the silica gel plate and the development solvent for the spotted material. Visualization of colored compounds is simple and the spots can be directly observed after development. Because most compounds are colorless; however, a visualization method is needed. The silica gel on the TLC plate is impregnated with a fluorescent material that glows under ultraviolet light.[16] A spot will interfere with the fluorescence and appear as a dark spot on a glowing background. While under the ultraviolet (UV) light, the spots can be outlined with a pencil to mark their locations and calculate the Rf value.
In vitro dissolution profile and drug release mechanism
The dissolution studies were performed using a USP1-type dissolution apparatus (Lab India DS 800 Mumbai), India. Accurately, 250 mg of GCV was weighed and filled in capsules and dissolution was performed in 900 mL of phosphate buffer (pH 7.4 as receptor media). The temperature was maintained at 37 ± 0.5°C, and the stirring speed employed was 100 rpm. Samples of 5 mL were withdrawn at various time points from each dissolution beaker and were replaced with fresh media maintained at the same temperature. The same procedure was adopted for the drug-excipient physical mixture as well. The withdrawn samples were filtered through a 0.2 µm nylon filter and analyzed at 254 nm using 6850 UV/Visible Spectrophotometer, Jenway. The release data were accordingly evaluated for drug percentage dissolved in the dissolution media.
Molecular docking
Docking was carried out using Auto Dock VINA 1.2.0 software.[17-19] The 2D structures of the molecules were downloaded from the PubChem database, and initial geometric optimization was carried out using the Gaussian software at the DFT/B3LYP level of theory with 6–311+G+basis et. The structures for docking were prepared using the AutoDock tools, and a blind docking was performed with a grid size 40Å*40Å*40Å.[17-19]
Molecular dynamics stimulations
The best docking pose generated from AutoDock VINA was taken as input for performing (molecular dynamics) MD stimulations. The GCV complexed with MCC and lactose were stimulated using the GROMACS 5.0 software,[20-23] and each 10 ns long with CHARMM 36 m[20-23] force field. TIP3 water model was used for each of the stimulations and equilibration was carried out at 303 K temperature. The various interactions and trajectories were analyzed using the GROMACS 5.0 software package.
RESULTS
Results of compatibility studies using FTIR spectroscopy
The FTIR spectra showed characteristic absorption bands of drug and excipients as it was confirmed from the literature and FTIR spectra of drug and mixture of drug and excipients were shown in the [Figure 1a-e]. The FTIR spectra of GCV are characterized by the absorption peaks at 3418.54 cm−1 (N-H stretch), 3318.15 cm−1 (O-H stretch), 3148.33 cm−1 (C-H stretch), 1685.18 cm−1 (C=O stretch), 1658.10 cm−1 (C=C stretch), 1571.87 cm−1 (C=N stretch), and 1364.89 cm−1 (O-H bending). Further, a comparative study for the FTIR bands of pure GCV and a physical mixture of different excipients was done and it is shown in [Table 1].
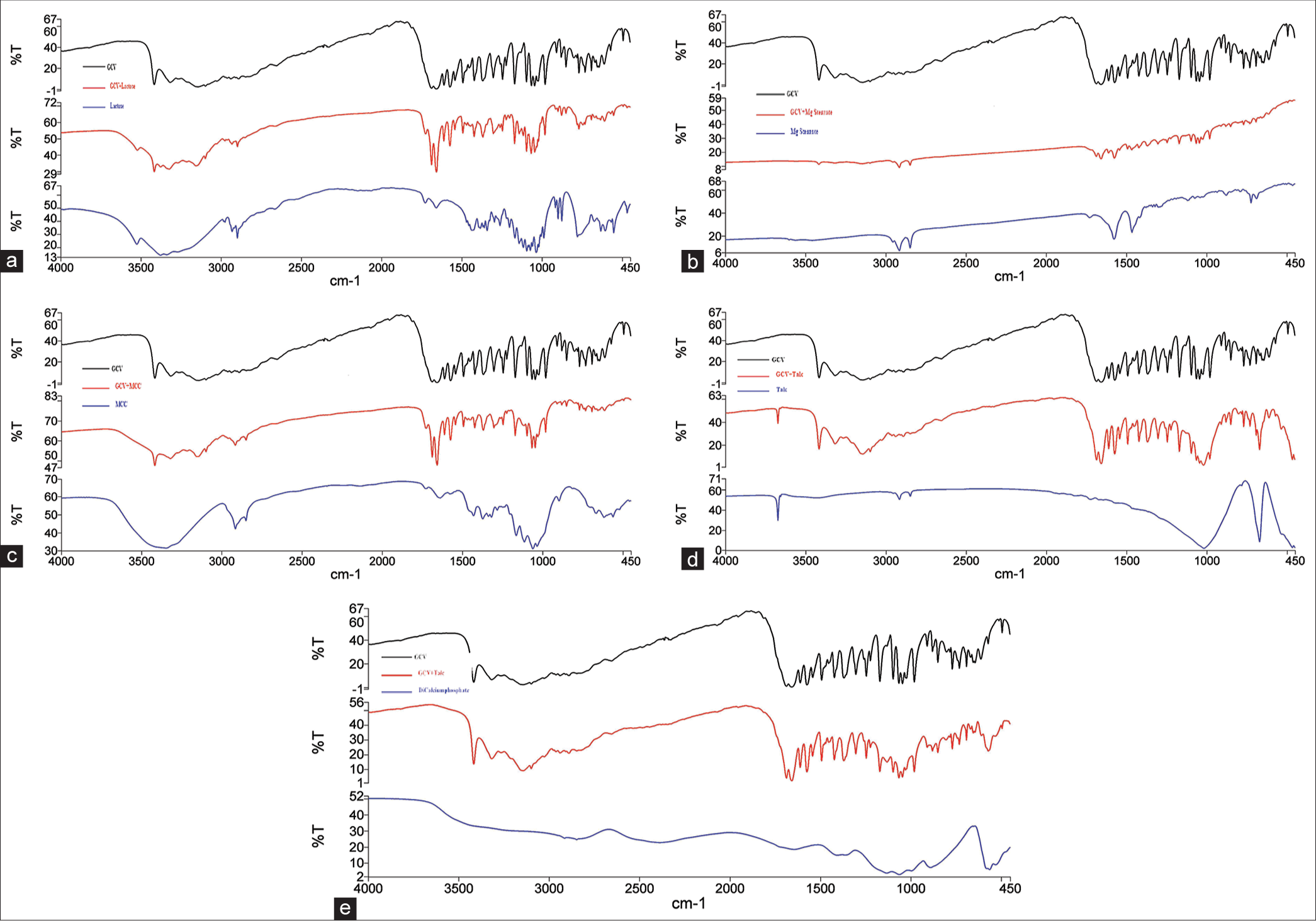
- (a) Fourier-transform infrared spectroscopy (FTIR) spectra of ganciclovir (GCV), Lactose and physical mixtures of GCV and Lactose. (b) FTIR spectra of GCV, Mg stearate, and physical mixtures of GCV and Mg- stearate. (c) FTIR spectra of GCV, microcrystalline cellulose (MCC), and physical mixtures of GCV and MCC. (d) FTIR spectra of GCV, talc, and physical mixtures of GCV and talc. (e) FTIR spectra of GCV, dicalcium phosphate, and physical mixtures of GCV and dicalcium phosphate.
| Functional group | GCV | GCV+LAC | GCV+MgS | GCV+MCC | GCV+TALC | GCV+DP |
|---|---|---|---|---|---|---|
| N-H stretch | 3418.54 | 3417.55 | 3422 | 3419.10 | 3419.02 | 3418.76 |
| O-H stretch | 3318.15 | 3329.00 | 3317.52 | 3318.37 | 3318.38 | 3317.89 |
| Aromatic C-H stretch | 3148.33 | 3156.40 | 3147.90 | 3152.37 | 3148.94 | 3148.45 |
| C=O stretch | 1685.18 | 1687.25 | 1687.34 | 1687.37 | 1685.80 | 1685.86 |
| Aromatic C=C stretch | 1658.10 | 1658.98 | 1658.56 | 1658.78 | 1658.63 | 1658.41 |
| Aromatic C=N | 1571.87 | 1573.76 | 1574.38 | 1573.54 | 1572.91 | 1572.85 |
| O-H bending | 1364.89 | 1367.77 | 1366.68 | 1367.94 | 1366.07 | 1365.74 |
FTIR: Fourier-transform infrared spectroscopy, GCV: Ganciclovir, LAC: Lactose, MCC: Microcrystalline cellulose, MgS: Magnesium stearate, DP: Dicalcium phosphate
Results of compatibility studies using 1H NMR spectroscopy
The 1H NMR of GCV exhibited signals with characteristic values of chemical shift (δ) such as 4.61 δ (CH2-CH-CH2), 5.45 δ (N-CH2O), 6.49 δ (CH-NH2), 7.81 δ (HetAr-NH2), and 10.65 δ (Ar-NH) The comparative analysis of chemical shifts of the GCV along with their excipients is shown in [Table 2].
| Chemical shift (ppm) | GCV | GCV+LAC | GCV+MgS | GCV+MCC | GCV+TALC | GCV+DP |
|---|---|---|---|---|---|---|
| CH2-CH-CH2 | 4.61 | 4.60 | 4.60 | 4.59 | 4.59 | 4.59 |
| N-CH2O | 5.45 | 5.44 | 5.43 | 5.40 | 5.44 | 5.44 |
| CH-NH2 | 6.49 | 6.48 | 6.47 | 6.48 | 6.47 | 6.48 |
| HetAr-NH2 | 7.81 | 7.80 | 7.79 | 7.79 | 7.80 | 7.80 |
| Ar-NH | 10.65 | 10.62 | 10.61 | 10.62 | 10.61 | 10.62 |
GCV: Ganciclovir, LAC: Lactose, MCC: Microcrystalline cellulose, MgS: Magnesium stearate, DP: Dicalcium phosphate, ppm: Parts per million
Results of compatibility studies using DSC
The DSC thermogram of GCV exhibited two different endothermic peaks at temperatures 238.82°C and 255°C, [Table 3] which were found to be comparable with the published literature. The DSC thermogram of pure GCV and pure GCV and physical mixture with excipients (lactose, MCC, magnesium stearate, talc, and dicalcium phosphate) is shown in [Figure 2a-f and Table 3].
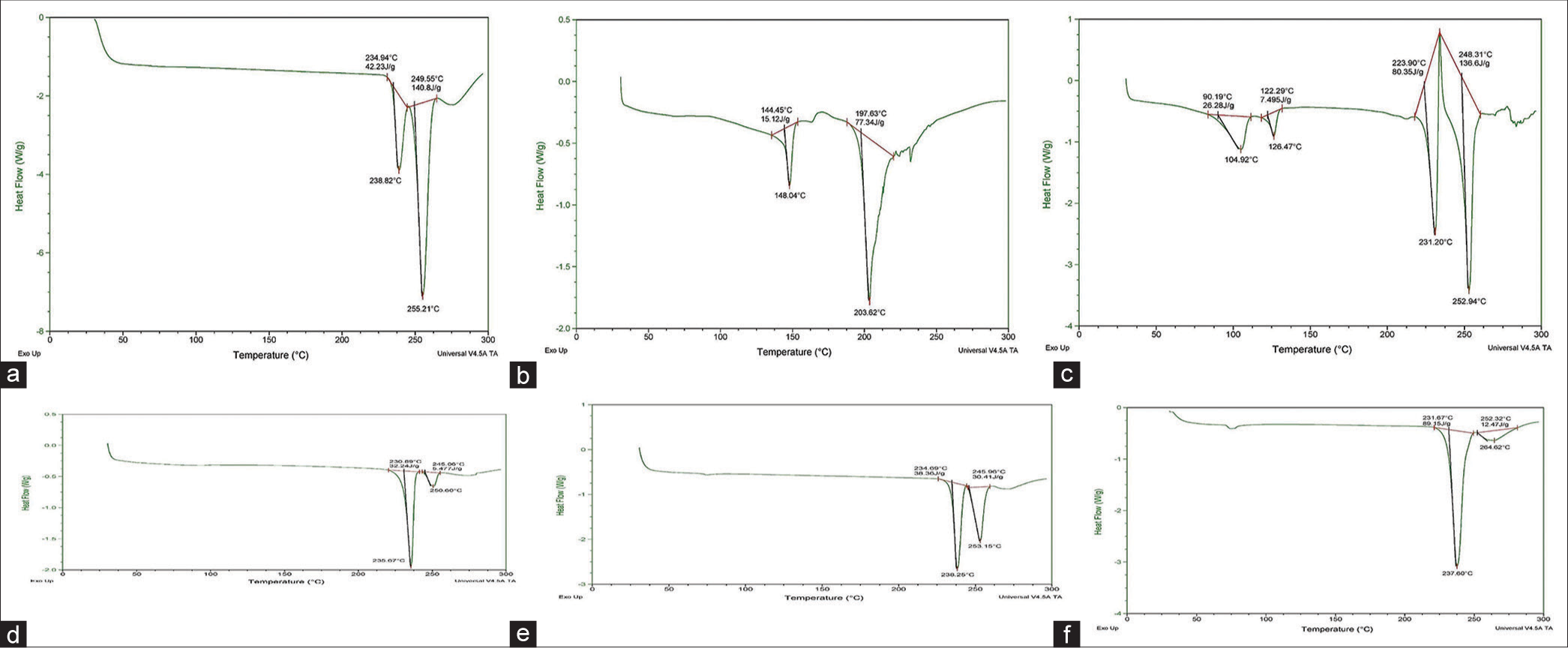
- (a) Differential scanning calorimetry (DSC) thermogram for ganciclovir (GCV). (b) DSC thermogram for physical mixtures of GCV and LAC. (c) DSC thermogram for physical mixtures of GCV and MgS. (d) DSC thermogram for physical mixtures of GCV and microcrystalline cellulose. (e) DSC thermogram for physical mixtures of GCV and TALC. (f) DSC thermogram for physical mixtures of GCV and DP. GCV: LAC: Lactose, MgS: Magnesium stearate, DP: Dicalcium phosphate.
| Sample | Drug-excipient ratio | Endothermic peaks |
|---|---|---|
| GCV | 100 | 238.82°C and 255°C |
| GCV-Lactose | 50:50 | 203.62°C |
| GCV-MCC | 50:50 | 235.67°C and 250.60°C |
| GCV-MgS | 50:50 | 231°C and 252°C |
| GCV-Talc | 50:50 | 238.25°C and 253.15°C |
| GCV-DP | 50:50 | 237.60°C and 264.62°C |
GCV: Ganciclovir, MCC: Microcrystalline cellulose, MgS: Magnesium stearate, DP: Dicalcium phosphate
Results of compatibility studies using TLC
The Rf value is used to quantify the movement of the materials along the plate. Rf is equal to the distance traveled by the substance divided by the distance traveled by the solvent. Its value is always between zero and one. The Rf value of GCV and physical mixtures of GCV with different excipients (lactose, magnesium stearate, MCC, talc, and dicalcium phosphate) were found to be 0.35.
Results of influence of hydrophobic and hydrophilic attribute of excipient over dissolution profile
The dissolution behavior of GCV for the physical mixtures containing different types of excipients is shown in [Figure 3]. The dissolution behavior of pure GCV and the physical mixtures containing different types of excipients were studied for 60 min. It was observed that GCV with talc showed at least 44.48% drug release for 60 min whereas GCV with MCC displayed a maximum 96% drug release for 60 min.
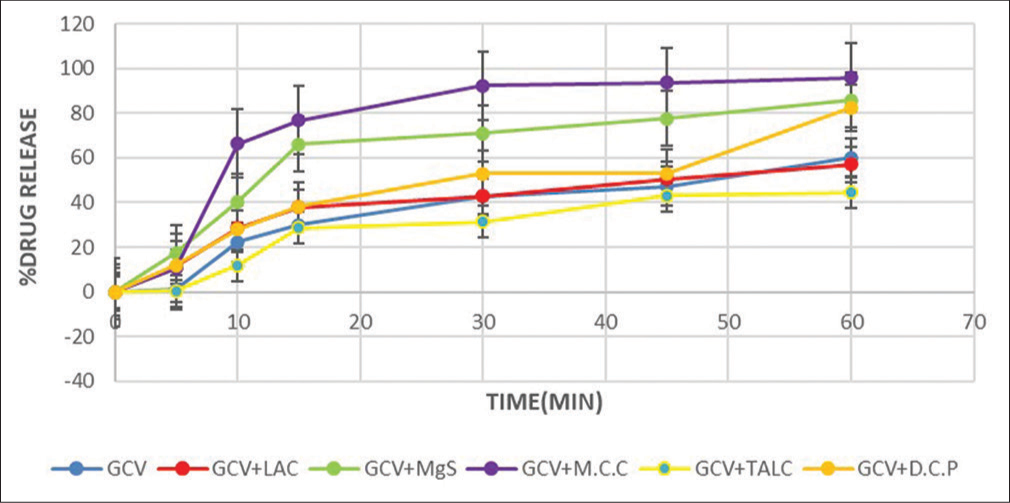
-
In vitro dissolution studies of ganciclovir (GCV) and physical mixtures of GCV and different excipients.
Results of molecular docking
The result of two docking studies, that is, GCV/MCC and GCV/LAC is shown in [Table 4] and the 3D pose of docked complexes GCV/MCC and GCV/LAC is shown in [Figure 4].
| S.No. | Complex name | Docking score |
|---|---|---|
| 1. | GCV/LAC | -2.7 |
| 2. | GCV/MCC | -2.4 |
GCV: Ganciclovir, LAC: Lactose, MCC: Microcrystalline cellulose
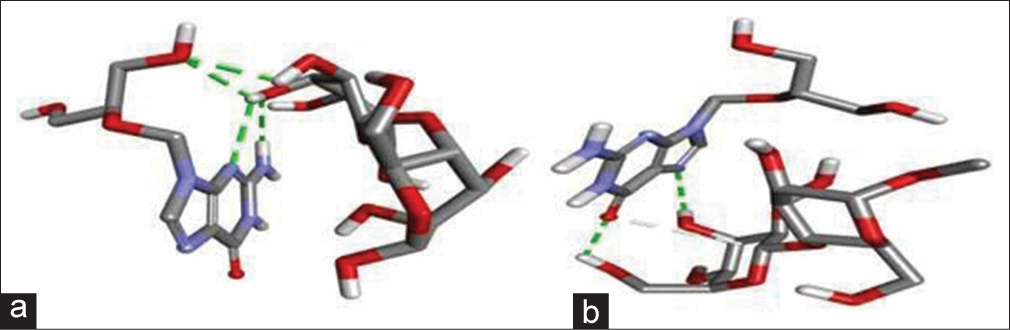
- (a) 3D docked pose of ganciclovir (GCV)/lactose (LAC), and (b) GCV/microcrystalline cellulose.
Docking scores for complexes of GCV/LAC and GCV/MCC were observed −2.7 and −2.4, respectively.
Results of molecular dynamics simulations
[Figure 5] signifies the analyses obtained from docking studies for 10 ns. From [Figure 5], it was observed that GCV/ LAC complex showed less deviations with less root mean square deviation (RMSD) value than GCV/MCC. Root mean square fluctuation (RMSF) plots displayed the movement of individual atoms of the drug in the mixture of the drug with the excipients.
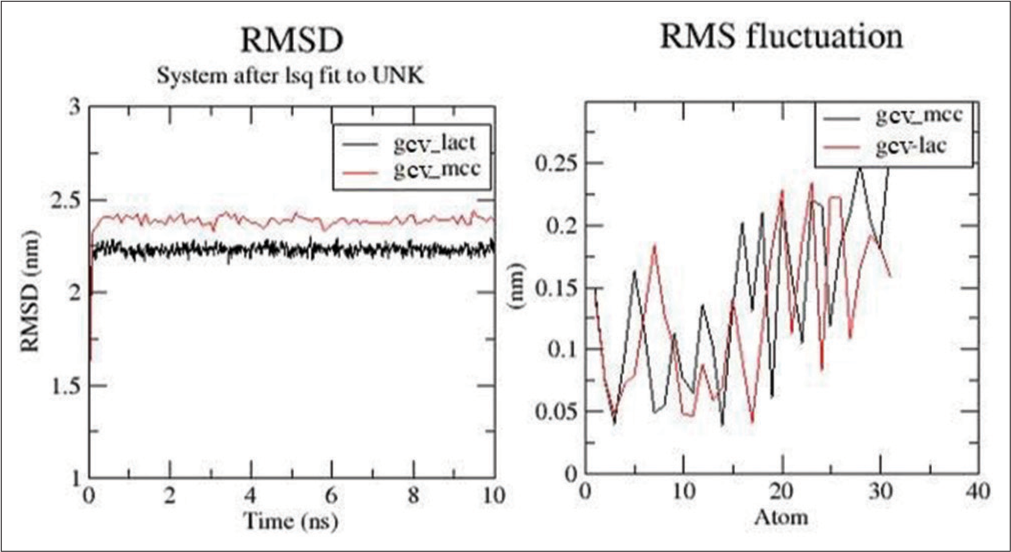
- Root mean square deviation (RMSD) and root mean square fluctuation (RMSF) plots for ganciclovir (GCV)/lactose (LAC) and GCV/microcrystalline cellulose.
[Figure 6] displayed the number of hydrogen bonds formed between the drug and excipient throughout the stimulation. The drug formed more hydrogen bonds with LAC in comparison to MCC.

- Hydrogen bonds for ganciclovir (GCV)/lactose (LAC) and GCV/microcrystalline cellulose (MCC) in each ps.
[Figure 6] designates the number of hydrogen bonds formed between the drug and excipient during the stimulation.
Solvent assessable surface area for both GCV/LAC and GCV/MCC are shown in [Figure 7] and their ranges were found to be 4.2–5 nm2.
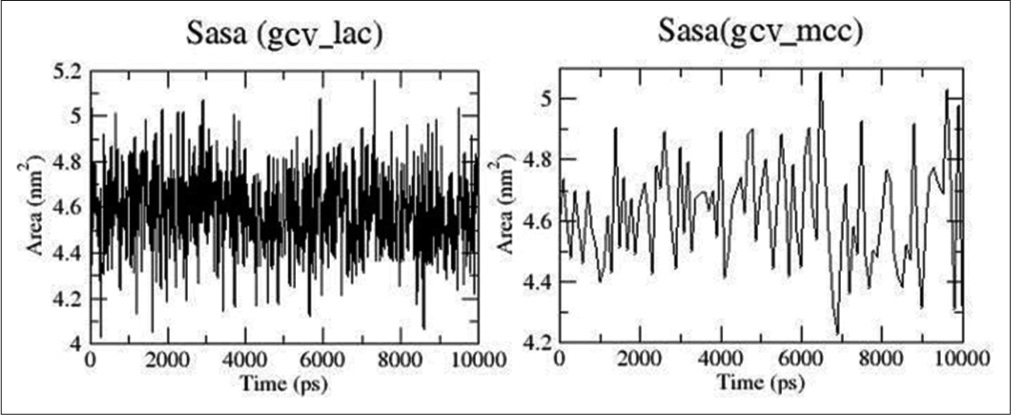
- Solvent assessable surface area plots for ganciclovir (GCV)/lactose (LAC) and GCV/microcrystalline cellulose (MCC).
The radius of gyration for the complex of the drug which was complexed with LAC and MCC was fluctuating in the range of 0.32–0.36 nm in both cases, as shown in [Figure 8]. The LJ, coulomb, and total interaction energies for both two systems were calculated and plotted on the graph [Figure 9]. The total energy of GCV/MCC and GCV/LAC is −41.1KJ and −33.3 KJ, respectively.

- Radius of gyration plots for ganciclovir (GCV)/lactose (LAC) and GCV/microcrystalline cellulose.

- Energy plots (Lennard jones, coulombic, and total) for ganciclovir (GCV)/lactose (LAC) and GCV/microcrystalline cellulose (MCC).
DISCUSSION
Compatibility studies using FTIR spectroscopy
The FTIR spectrum displayed characteristic absorption bands of the drug and physical mixtures of the drug with excipients, as shown in [Figure 1a-f and Table 1]. The FTIR spectrum of drug GCV was characterized by absorption peaks at 3418.54 cm−1 (N-H stretch), 3318.15 cm−1 (O-H stretch), 3148.33 cm−1 (C-H stretch), 1685.18 cm−1 (C=O stretch), 1658.10 cm−1 (C=C stretch), 1571.87 cm−1 (C=N stretch), and 1364.89 cm−1 (O-H bending). The characteristic absorption bands of pure drug and physical mixture are shown in [Table 1 and Figure 1a-f]. The characteristic bands of functional groups at 3418.54 cm-1 (N-H stretch), 3318.15 cm-1 (O-H stretch), 3148.33 cm−1 (C-H stretch), 1685.18 cm−1 (C=O stretch), 1658.10 cm−1 (C=C stretch), 1571.87 cm−1 (C=N stretch), and 1364.89 cm−1 (O-H bending) in FTIR spectra of drug GCV were retained in FTIR spectra of the physical mixture of lactose, magnesium stearate, MCC, talc, and dicalcium phosphate [Table 1 and Figure 1a-f] and this signified that there were no incompatibilities between drug and physical mixtures of drug with excipients. The obtained results from FTIR showed that the GCV was compatible with all used excipients such as lactose, magnesium stearate, MCC, talc, and dicalcium phosphate.
Compatibility studies using 1H NMR spectroscopy
Several studies have been reported using 1H NMR to identify any interactions in drugs with excipients. The comparative analysis for chemical shifts of the drug GCV and physical mixtures is shown in [Table 2]. Most of the important signals of GCV such as CH2-CH-CH2 (4.61); N-CH2O (5.45), CHNH2 (6.49), Het Ar-NH2 (7.81), and Ar-NH (10.65) were present nearly at the same position in the spectra of the physical mixture of GCV and excipients such as lactose, magnesium stearate, MCC, talc, and dicalcium phosphate [Table 2]. No significant shifting of chemical shift was observed which displayed that there were no observed interactions between the drug and excipient. Thus, it was predicted that GCV was compatible with all used excipients such as lactose, magnesium stearate, MCC, talc, and dicalcium phosphate.
Compatibility studies using DSC
The DSC thermogram of pure GCV reflected two sharp endothermic peaks at 238.82°C and 255°C [Figure 2a and Table 3]. The DSC thermogram of the physical mixture of GCV with lactose is shown in [Figure 2b]. In the thermogram of the physical mixture of GCV and lactose, the endothermic peak corresponding to GCV was observed at 203.62°C. It was observed that the peaks corresponding to pure GCV were not retained when it was masked through tlc, and it was concluded that the spot of GCV and physical mixture of lactose with GCV was found to be in the same position. We predicted that there were no chemical incompatibilities between GCV and lactose. The DSC thermogram of the physical mixture of GCV with magnesium stearate was studied [Figure 2c]. In the thermogram of the physical mixture of GCV with magnesium stearate, the endothermic peaks corresponding to GCV were observed at 231°C and 252°C. The peaks corresponding to pure GCV were retained with slight modifications and it demonstrated that there was no chemical interaction between GCV and magnesium stearate. In the DSC thermogram of the physical mixture of GCV with MCC, the endothermic peak corresponding to GCV was observed at 235.67°C and 250.60°C [Figure 2d]. It was found that the peaks corresponding to pure GCV were retained with slight modifications. Further, it was concluded that there was no chemical interaction between GCV and MCC. The DSC thermogram of the physical mixture of GCV with talc reflected two sharp endothermic peaks at 238.82°C and 255°C. In the thermogram of the physical mixture of GCV with talc, the endothermic peak corresponding to GCV was observed at 238.25°C and 253.15°C [Figure 2e]. It was observed that the peak corresponding to pure GCV was observed at the same position with slight modifications. We concluded that there was no chemical interaction between GCV and talc. In the DSC thermograms of the physical mixture of GCV and dicalcium phosphate, the endothermic peak corresponding to GCV was observed at 237.60°C and 264.62°C [Figure 2f]. It showed that the peaks corresponding to pure GCV were retained with slight modifications and it was concluded that there was no chemical interaction between GCV and dicalcium phosphate.
Compatibility studies using TLC
The Rf value is used to enumerate the movement of the materials along the plate. Rf is equivalent to the distance traveled by the substance divided by the distance traveled by the solvent. Its value is always between zero and one. The Rf value of GCV and physical mixtures with different excipients (lactose, magnesium stearate, MCC, talc, and dicalcium phosphate) was found to be 0.35. The spot of GCV and GCV with different excipients was visible at the same position. From this, it was decided that no interaction was observed among the GCV and all physical mixtures of different excipients.
Influence of hydrophobic and hydrophilic attribute of excipient over dissolution profile
The dissolution behavior of GCV for the physical mixtures containing different types of excipients is shown in [Figure 3]. The GCV drug release is different for different excipients. The amount of drug release at 10 min was found to be quite similar for two types of excipients (lactose and dicalcium phosphate) which were found to be 28.68% and 28.2%, respectively. The amount of drug released in the mixture of GCV and magnesium stearate was found to be 40.52%. At 30 min., the amount of drug released in the mixture of GCV and lactose was observed to be quite similar to that of pure drug, that is, 42.92% and 42.76%, respectively. The physical mixture of GCV and MCC showed a maximum amount of drug release in a shorter period. On the other hand, the physical mixture of GCV and talc showed the minimum amount of drug release compared to other excipients. This may be attributed to the hydrophobic nature of talc. It was observed that the drug release from the physical mixtures containing different excipients was influenced by the hydrophobicity and hydrophilicity of the excipients used.
Molecular docking
Molecular docking is an established in silico methodology that enables identification of drug interactions at the molecular level. Docking studies were performed to find out the possible interactions of the excipients with the GCV. The docking study also gives insights into the lowest energy conformations. Two docking studies, that is, GCV/MCC and GCV/LAC, were carried out and the lowest energy conformer of each of the two experiments was taken as the most suitable conformer. The docking score of each complex is shown in [Table 4] and the 3D pose of the docked complexes GCV/MCC and GCV/LAC is shown in [Figure 4]. Docking scores for complexes of GCV/LAC and GCV/MCC was observed −2.7 and −2.4, respectively, and this docking study showed that the number of interactions were more with GCV and LAC (four nos.) in comparison to GCV and MCC (two nos.). This interaction supported the drug release of GCV with MCC which was higher than GCV with LAC.
Molecular dynamics simulations
The GCV drug was complexed with LAC and MCC, and the best pose obtained from docking was used to run the 10 ns stimulations. [Figure 5] represents the analyses obtained from docking studies. [Figure 5] indicated that the GCV/LAC complex was showing less deviations with less RMSD value than GCV/MCC. Both the complexes were stable throughout the stimulations. RMSF plots showed the movement of individual atoms of the drug which was complex with the excipients. The RMSF plot designated considerable movement of individual atoms. [Figure 6] showed the number of hydrogen bonds formed between the drug and excipient throughout the stimulation. It was evident that the drug was forming more hydrogen bonds with LAC and was more stable compared to the number and stability of hydrogen bonds between GCV and MCC. Solvent assessable surface area for both GCV/LAC and GCV/MCC is shown in [Figure 7] and was found to be fluctuating in the range of 4.2–5 nm2. The drug was thought to have the same assess to the solvent when combined with both LAC and MCC.
The radius of gyration of the drug which was complexed with LAC and MCC was plotted on the graph and the values fluctuated between the ranges of 0.32–0.36 nm in both cases, as shown in [Figure 8]. The high fluctuation suggested that the drug was not very compact in the system.
The LJ, coulomb, and total interaction energies of the two systems were calculated and plotted on the graph [Figure 9]. Here, both systems show similar LJ interactions, while the coulombic interactions were varying. The low total energy of GCV/MCC (−41.1 KJ) to GCV/LAC (−33.3 KJ) suggested that MCC was having stronger interaction with the drug compared to LAC. The radius of gyration plots for both GCV/LAC and GCV/MCC is shown in [Figure 8].
CONCLUSION
The compatibility studies of GCV were studied by FTIR, DSC, and 1H NMR and it was observed that there was no significant deviation in the characteristic peaks in infrared spectroscopy and 1H NMR signals. Based on the above discussion, it was concluded that GCV should be compatible with all types of excipients used in this study. Further to see the compatibility studies, DSC was performed using individual drugs and in combination with excipients in the form of physical mixtures. GCV was found to be compatible with all types of excipients but it was masked through TLC that all the spots in the individual drug sample and the physical mixtures containing drugs and excipients were observed to be at the same position which showed that there were no interactions. Further, it was concluded that GCV should be compatible with all types of excipients. The drug dissolution behavior of the GCV and the physical mixture of MCC with the drug showed a maximum amount of drug release in a short period of time. The release of drug from the physical mixtures containing different excipients was influenced by the dual effects of hydrophobicity and hydrophilicity of the different excipients.
The % drug release at 10 min was found to be quite similar for lactose (28.68%) and dicalcium phosphate (28.2%). A physical mixture of GCV with MCC displayed a maximum amount (96%) of the drug for 60 min. On the other hand, the physical mixture of GCV with talc showed a minimum amount (44.48%) of drug release compared to other excipients’ drug release for 60 min. Docking studies were done to find out the possible interactions of excipients with the drug molecule. In GCV, the lactose complex is showing less deviation with less RMSD value as compared with MCC complex. The lactose complex having more hydrogen bonding and was more stable as compared with the MCC complex. GCV indicates that the total energy of MCC complex is less than that of the lactose complex. This indicates that GCV is more soluble when combined with the microcrystalline complex.
Acknowledgments
The authors are thankful to DST-FIST Lab of University Institute of Pharmaceutical Sciences for providing different instruments to do experiments.
Ethical approval
Not applicable.
Declaration of patient consent
Patient’s consent not required as there are no patients in this study.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Financial support and sponsorship
Nil.
References
- Drug-excipient compatibility studies: First step for dosage form development. Pharma Innov. 2015;4:14-20.
- [Google Scholar]
- An overview of pharmaceutical excipients: Interactions and incompatibilities in dosage form development. Pharm Res J. 2022;1:65-72.
- [Google Scholar]
- Excipient applications in formulation design and drug delivery. Germany: Springer; 2015:1-10.
- [CrossRef] [Google Scholar]
- Drug-excipient compatibility assessment of solid formulations containing meloxicam. Eur J Pharm Sci. 2018;112:146-51.
- [CrossRef] [Google Scholar]
- Development of an analytical UHPLC method for the estimation of cyclosporine in SMEDDS and protein binding. Am J Mod Chromatogr. 2017;4:1-12.
- [CrossRef] [Google Scholar]
- Drug-excipient compatibility studies in formulation development: Current trends and techniques United States: St. John Fisher University; 2015. p. :9-15.
- [Google Scholar]
- Drug-excipient compatibility screening--role of thermoanalytical and spectroscopic techniques. J Pharm Biomed Anal. 2014;87:82-97.
- [CrossRef] [Google Scholar]
- Interaction and compatibility studies in the development of olmesartan medoxomil and hydrochlorothiazide formulations under a real manufacturing process. Pharmaceutics. 2022;14:424.
- [CrossRef] [Google Scholar]
- Application of attenuated total reflectance FTIR spectroscopy to the analysis of mixtures of pharmaceutical polymorphs. Int J Pharm. 1998;163:157-66.
- [CrossRef] [Google Scholar]
- DSC supported by factor analysis as a reliable tool for compatibility study in pharmaceutical mixtures. J Therm Anal Calorim. 2019;138:4531-9.
- [CrossRef] [Google Scholar]
- Application of differential scanning calorimetry to the study of drug-excipient compatibility. Thermochim Acta. 1996;285:337-45.
- [CrossRef] [Google Scholar]
- An evaluation of the potential of NMR spectroscopy and computational modelling methods to inform biopharmaceutical formulations. Pharmaceutics. 2018;10:165.
- [CrossRef] [Google Scholar]
- Comprehensive assessment of protein and excipient stability in biopharmaceutical formulations using 1H NMR spectroscopy. ACS Pharmacol Transl Sci. 2021;4:288-95.
- [CrossRef] [Google Scholar]
- Assignment of 1H nuclear magnetic resonance visible polyunsaturated fatty acids in BT4C gliomas undergoing ganciclovir-thymidine kinase gene therapy-induced programmed cell death. Cancer Res. 2003;63:3195-201.
- [Google Scholar]
- Thin-layer chromatography: A modern practical approach United Kingdom: Royal Society of Chemistry; 2007.
- [Google Scholar]
- AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455-61.
- [CrossRef] [PubMed] [Google Scholar]
- PubChem substance and compound databases. Nucleic Acids Res. 2016;44:D1202-13.
- [CrossRef] [PubMed] [Google Scholar]
- Using AutoDock with AutoDockTools: A tutorial California, USA: The Scripps Research Institute Molecular Graphics Laboratory; 2006.
- [Google Scholar]
- GROMACS: Fast, flexible, and free. J Comput Chem. 2005;26:1701-18.
- [CrossRef] [PubMed] [Google Scholar]
- CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat Methods. 2017;14:71-3.
- [CrossRef] [PubMed] [Google Scholar]
- The Lennard-Jones potential: When (not) to use it. Phys Chem Chem Phys. 2020;22:10624-33.
- [CrossRef] [PubMed] [Google Scholar]




