
Translate this page into:
Selenium-Dependent Read Through of the Conserved 3’-Terminal UGA Stop Codon of HIV-1 nef


-
Received: ,
Accepted: ,
How to cite this article: Premadasa L, Dailey G, Ruzicka JA, Taylor EW. Selenium-dependent read through of the conserved 3’-terminal UGA stop codon of HIV-1 nef. Am J Biopharm Pharm Sci 2021;1:1.
Abstract
Objectives:
The HIV-1 nef gene terminates in a 3’-UGA stop codon, which is highly conserved in the main group of HIV-1 subtypes, along with a downstream potential coding region that could extend the nef protein by 33 amino acids, if readthrough of the stop codon occurs. It has been proposed that antisense tethering interactions (ATIs) between a viral mRNA and a host selenoprotein mRNA are a potential viral strategy for the capture of a host selenocysteine insertion sequence (SECIS) element. This mRNA hijacking mechanism could enable the expression of virally encoded selenoprotein modules, through translation of in-frame UGA stop codons as selenocysteine (Sec). Here, our aim was to assess whether readthrough of the 3’-terminal UGA codon of nef occurs during translation of HIV-1 nef expression constructs in transfected cells, and whether selenium-based mechanisms might be involved.
Material and Methods:
To assess UGA codon readthrough, we used fluorescence microscopy image analysis and flow cytometry of HEK 293 cells transfected with full length HIV-1 nef gene expression constructs including the 3’-UGA stop codon and a predicted thioredoxin reductase 1 (TXNRD1) antisense region spanning the UGA codon, engineered with a downstream in-frame green fluorescent protein (GFP) reporter gene. These were designed so that GFP can only be expressed by translational recoding of the UGA codon, that is, if the UGA codon is translated as an amino acid or bypassed by ribosomal hopping. To assess readthrough efficiency, appropriate mutant control constructs were used for 100% and 0% readthrough. We used anti-TXNRD1 siRNA to assess the possible role of the proposed antisense interaction in this event, by knockdown of TXNRD1 mRNA levels.
Results:
UGA stop codon readthrough efficiency for the wild-type nef construct was estimated by flow cytometry to be about 19% (P < 0.0001). siRNA knockdown of TXNRD1 mRNA resulted in a 67% decrease in GFP expression in this system relative to control cells (P < 0.0001), presumably due to reduced availability of the components involved in selenocysteine incorporation for the stop codon readthrough (i.e. the TXNRD1 SECIS element). Addition of 20 nM sodium selenite to the media enhanced stop codon readthrough in the pNefATI1 plasmid construct by >100% (P < 0.0001), that is, more than doubled the amount of readthrough product, supporting the hypothesis that selenium is involved in the UGA readthrough mechanism.
Conclusion:
Our results show that readthrough of the 3’-terminal UGA codon of nef occurs during translation of HIV-1 nef expression constructs in transfected cells, that this is dependent on selenium concentration, and the presence of TXNRD1 mRNA, supporting the proposed antisense tethering interaction.
Keywords
HIV-1
nef
Thioredoxin reductase 1
Selenium
Antisense

INTRODUCTION
Taylor et al. previously proposed the possibility of viral RNA/ host mRNA antisense interactions as a gain-of-function strategy for viruses, through the tethering of a host mRNA containing functional structural or sequence elements.[1] That study focused on two viral genes, the nucleoprotein (NP) of the highly pathogenic Ebola Zaire strain (EBOV) and the HIV-1 nef gene, both of which terminate in highly conserved UGA stop codons, and both of which have, in close proximity to the UGA codon, extensive regions of antisense complementarity to the mRNA of an isoform of the mammalian selenoprotein thioredoxin reductase (TXNRD), specifically TXNRD3 in the case of the EBOV NP gene, and TXNRD1 in the case of HIV-1 nef. It was proposed that these regions could provide the basis for “antisense tethering interactions” (ATI), and that in both cases the functional element being targeted by viral capture is the selenocysteine insertion sequence (SECIS) element located in the 3’ untranslated region (3’-UTR) of all human TXNRD mRNAs.[2] This structural element is essential for translational recoding of in-frame UGA stop codons as selenocysteine (Sec), an amino acid whose presence is the defining feature of selenoproteins.[3] Thus, it seems unlikely to be a coincidence that the two genes in question, from very different RNA viruses, should both show evidence of antisense targeting of a human selenoprotein mRNA, that it is a TXNRD isoform in both cases, in regions of the viral genome that prove to overlap or be within a few hundred bases on either side of a highly conserved in-frame UGA codon, recoding of which as Sec would lead to an extended selenoprotein isoform of the respective proteins (NP and nef), the sequence features of which are also conserved, even past the UGA stop codon.
In mammalian TXNRD enzymes, the Sec residue encoded by the UGA is geminal to a Cys residue at the C-terminal end of the protein, the Cys-Sec pair forming the C-terminal redox center of TXNRD enzymes.[2] As we have pointed out previously, this is strikingly similar to the situation in the HIV- nef gene, which has a conserved C terminal Cys residue, followed immediately by the UGA stop codon, thus potentially forming a TXNRD-like Cys-Sec redox pair if the UGA codon could be translated as Sec.[4]
Selenoproteins encoded in the genomes of bacteria, eukaryotes and archaea all encode Sec using the UGA codon, which otherwise, and far more commonly, serves as a stop codon.[5] To incorporate Sec into polypeptides, these organisms all use variants of a complex co-translational mechanism, a key feature of which is the involvement of a characteristic RNA stem-loop structure. In the mammalian system, this structure is the SECIS element and is usually located in the 3’ UTR of the selenoprotein mRNA. Various protein factors, including SECIS binding protein 2 and a special elongation factor, EFSec, work in conjunction with the SECIS element to bind tRNASec at the ribosome, suppress the termination factor, and thereby enable Sec incorporation at the UGA codon, preventing it from acting as a stop codon [Figure 1, upper panel].

- (a and b) Proposed mechanism of Sec incorporation into viral proteins via hijacking of a SECIS element from a tethered host selenoprotein mRNA. (Reproduced from Figure 3 of Taylor et al., 2016[1]). Both panels show schematic ribosomes with two bound tRNAs, one carrying the Sec, the other a growing peptide chain shown as colored circles. The upper Panel A shows the established role of the SECIS element in the 3’-UTR region in the mechanism of insertion of Sec during mammalian selenoprotein biosynthesis.[5,10] The crossed-out STOP indicates that the SECIS mechanism illustrated here serves to override the usual termination function of an in-frame UGA codon. Panel B shows how the HIV-1 mRNA could hijack the host SECIS element via antisense tethering interactions (ATI) to recode UGA as Sec, to synthesize viral selenoproteins such as the HIV-1 encoded GPx.[7] ATI-1 is a predicted interaction spanning the highly conserved 3’ UGA codon of the nef gene. ATI-2 is a second shorter antisense region consisting of 13 consecutive bases near the end of viral mRNA.
Given the universality of UGA codon use for Sec, it should not be surprising that viruses might also exploit this mechanism and benefit by the ability to synthesize alternate isoforms of certain viral proteins, for example, by extending a protein through readthrough of a stop codon. The latter is actually a general strategy of some viruses, and recoding of UGA as Sec is only one special case of readthrough suppression, for which there are diverse mechanisms that can result in conventional amino acids being incorporated at any of the three stop codons, depending on the context. For example, murine leukemia virus can only express its pol gene through translational readthrough involving the insertion of a glutamine at the position of a UAG stop codon that separates the gag and pol reading frames, an event that occurs at low but adequate efficiency (5%).[6] This has evolved to give an appropriate 20:1 molar ratio in the quantity of gag structural proteins versus the enzymes encoded in the pol gene.
In addition to a potential selenoprotein isoform of nef associated with its 3’-UGA codon, based largely on computational analyses of the genomic structure of HIV-1, we have also previously identified several regions overlapping known HIV genes that potentially encode selenoprotein modules expressed by ribosomal frame shifting, including one that is a homologue of glutathione peroxidase (GPx),[7] and one with similarities to the transcription factor NF-κB, the primary cellular on/off switch for HIV.[4,8] The HIV-1 GPx was cloned and expressed using a human SECIS element, and was found to encode functional GPx activity,[7] and to protect transfected cells against oxidant-induced apoptosis.[9]
However, neither we nor others were ever able to identify a functional SECIS element encoded by an RNA virus, which engendered considerable skepticism in regard to our findings, for example, regarding the HIV-1 encoded GPx. The missing piece of the puzzle was finally provided by our 2016 hypothesis,[1] which is presented visually in the lower panel of Figure 1. The central idea is that a virus would not need its own SECIS element if it could hijack one from the host, through the mRNA tethering mechanism outlined above. Since SECIS elements have the ability to function even when carried by a separate mRNA,[10] it is highly probable that the presence of a SECIS in a tethered mRNA could also serve to decode certain in-frame UGA codons as Sec on the tethering viral RNA, particularly if this interaction was the result of a coevolutionary process and structural and mechanistic features had been optimized by evolutionary selection.
Our preliminary support for this hypothesis in the case of the Ebola NP and HIV-1 nef genes was through a combination of computational analyses and confirmation of the antisense interactions at the DNA level, through gel shift assay of the specific fragments that were predicted to be involved in the core antisense interaction (what we called the ATI1 region), shown as interacting RNA secondary structures in Figure 1 of Taylor et al.[1] The aim of the current study was to assess in vitro the predicted outcome of that interaction in the case of the nef gene, that is, the potential for formation of a C-terminal extended isoform of nef, and to seek evidence of a role for TXNRD1 mRNA in the efficiency of that readthrough.
To that end, we designed three nef expression constructs based on a green fluorescent protein (GFP) expression vector that included the complete nef coding region inserted upstream of GFP. In between the nef and GFP coding regions were either one or both of the predicted antisense regions: ATI1, spanning the nef 3’-UGA codon (pNefATI1 construct), and ATI2, which is further downstream in the 3’ end of the viral RNA (pNefATI2 construct, which contained both the ATI1 and ATI2 regions). These plasmids were designed so that in the “wild-type” pNefATI1 and pNefATI2 constructs, readthrough translation or bypassing of the nef 3’-UGA would enable translation of the downstream in-frame GFP domain. The third plasmid (pNefATIstop) was similar to pNefATI2, except for the presence of two in-frame stop codons (TAA and TAG) between the ATI1, ATI2, and GFP regions, to serve as a negative control by making GFP expression impossible even if UGA readthrough had occurred.
In brief, the results reported here exploit that set of nef ATI constructs, along with anti-TXNRD1 siRNA and appropriate control siRNAs, to demonstrate that readthrough of the nef 3’-UGA stop codon occurs with significant efficiency (~19%), is selenium-dependent, being more than doubled by the addition of 20 nM sodium selenite to cell culture media, and is significantly reduced, by about 67%, in the presence of an anti-TXNRD1 siRNA. These results unambiguously confirm the existence of an extended isoform of nef formed by translational stop codon readthrough, and are consistent with the hypothesis that TXNRD1 may play a role in the readthrough through the proposed antisense mechanism.
MATERIAL AND METHODS
Engineering of HIV-1 nef expression vector constructs designed to monitor UGA stop codon readthrough
To investigate the potential read-through of the UGA stop codon at the end of HIV-1 nef a set of three plasmid vectors was constructed, each containing a GFP reporter gene downstream of the complete HIV-1 nef coding region, including most of the 3’-LTR. The nef gene and LTR were obtained by PCR from the infectious molecular clone pNL4-3 (NIH AIDS Reagent Program 114; GenBank AF324493) and were ligated into the parent mammalian expression vector pAcGFP1-N3 (Takara Bio USA) such that GFP is expressed only if readthrough of the terminal UGA occurs. Primer design incorporated 5’ NheI and 3’ BamHI restriction sites into each of the three amplicons. To access GFP, an in-frame TAA stop codon at position 9504, between the two ATI regions, was mutated to CAA. The plasmids, shown schematically in Figure 2, were (1) pNefATI1, which contained nef and only the first of the two putative ATI regions, (2) pNefATI2, in which the second tethering region was additionally included, and (3) pNefATIstop, a double-stop negative control in which the stop codon at 9504 is retained and a second in-frame stop codon (TAG) is introduced just past the end of ATI2 but upstream of GFP, blocking formation of GFP even if readthrough of the UGA occurs. The primers (Integrated DNA Technologies, Inc, Coralville, IA; shown 5’ → 3’) included the same forward primer CAG GGC TAG CAA AGG ATT TTG CTA TAA CAT GGG TGG CAA G for all 3 constructs and reverse primers TGC AGG ATC CGA GGG CTC GCC ACT CC (pNefATI1), CCA GAG GGA TCC AGT ACA GGC AAA AAG CAG CTG CTT GTA TGC AGC ATC (pNefATI2), and GAG GGA TCC ACT ACA GGC AAA AAG CAG CTG CTT ATA TGC AGC ATC (pNefATIstop). Proper assembly of the vector constructs was verified by sequencing (Eurofins Genomics, Louisville, KY).

- Schematics for the plasmid inserts used to assess nef 3’-UGA readthrough. Regions with antisense complementarity to TXNRD1 mRNA are shown crosshatched as ATI1 and ATI2. Naturally occurring (TGA and TAA) or engineered (TAG) in-frame stop codons and a CAA mutant of the wild-type HIV TAA are indicated.
Selenium dependence of stop codon readthrough
To assess the influence of selenium on the readthrough of the terminal UGA codon in nef and optimize its concentration in the culture growth media, HEK 293T cells (NIH AIDS Reagent Program catalog number 103) were seeded (2 × 104/well) in a black clear-bottom 96-well plate (Costar 3603) and incubated for 24 h in DMEM supplemented with 10% fetal bovine serum at 37°C, 5% CO2 atmosphere, after which the media were replaced with media enriched with sodium selenite (0, 20, 50, or 80 nM). After 48 h the cells were transfected with pNefATI1 using lipofectamine 2000 (Invitrogen) by addition 50 μL of OptiMEM I (Gibco) containing 0.2 μg of plasmid DNA complexed with 0.3 μL of cationic lipid to each well. Incubation at 37°C was continued for 12 h and the media were replaced with fresh Se-enriched media. Four days post-transfection fluorescence was measured using a BioTek Synergy Mx microplate reader with excitation at 475 nm and emission at 505 nm.
Transfection of HEK 293T cells with pNefATI constructs
HEK 293T cells were transfected with each of the pNefATI plasmids using the protocol described above, except that all incubations contained 20 nM selenium (deemed the optimal concentration) after the initial 24 h, Se-free period. The expression of GFP was followed using an EVOS cell imaging system. Fluorescence images were taken at ten different well locations for each treatment and the average ± SD of the intensity was calculated using NIH ImageJ software.[11]
Flow cytometry analysis
HEK 293T cells were seeded in a six well plate (5 × 105/well) in 1 ml of media enriched with 20 nM selenium and incubated for 12 h. The cells were transfected with pNefATI1 or the parent pAcGFP1-N3 plasmid by addition of a complex of 0.5 μg plasmid DNA and 3.5 μL lipofectamine in 100 μL OptiMEM I. After 12 h, the media was replaced with fresh media (20 nM Se) and the cells were incubated for 4 days at 37°C. Cells were trypsinized, washed with 1% PBS, and resuspended in 400 μl of 0.1% BSA in PBS. GFP expression was evaluated using a BD FACSAria III flow cytometer with FACS DIVA version 6.1.3 software.
siRNA knockdown of TXNRD1 mRNA
TXNRD siRNA (Life Technologies 4390824) and the Ambion Silencer siRNA transfection II kit (Life Technologies AM1631) were used for the knockdown of TXNRD1 mRNA. Transfection of HEK 293 cells with pNefATI1 + siRNA was optimized using varying volumes of the two transfection agents supplied in the Silencer kit, siPORT Amine and siPORT. All incubations used growth media enriched with 20 nM Se.
To initiate an experiment, siPORT Amine (0.6 μL + 25 μL OptiMEM I/well) was mixed with pNefATI1 (0.2 μg + 25 μL OptiMEM I/well). Anti-TXNRD1 siRNA was reconstituted in nuclease-free water to a concentration of 20 μM and for each well 0.75 μL was diluted to a final volume of 25 μL in OptiMEM I. Aliquots (equivalent to 0.75 μL/well) of GADPH siRNA and scrambled siRNA (negative control) from the Silencer transfection kit were similarly diluted in OptiMEM I to a final total volume of 25 μL/well. The DNA/ siPORT Amine complex solution (50 μL/well) and siRNA solutions (25 μL/well) were combined and 75 μL aliquots were placed in each well of a 24 well culture plate. Control wells contained 75 μL of OptiMEM with or without siPORT Amine and no nucleic acid. HEK 293 cells (5 × 105 in 425 μL media) were added to each well. The final concentration of siRNA was 30 nM. Plates were incubated at 37°C for 72 h. GFP expression was imaged using the EVOS system and fluorescence intensity was calculated from ten images of each well using the NIH ImageJ software. An identical 24 well plate was prepared in tandem and used for qPCR analysis to quantify the siRNA knockdown of TXNRD1.
Quantitating TXNRD1 mRNA knockdown using RT-PCR
To confirm that the commercial anti-TXNRD1 siRNA was effective in our system, the cells in all treatment and control wells were harvested and processed to isolate total RNA (SV Total RNA Isolation System, Promega Corp., Madison, WI). cDNA was synthesized using 300 ng of RNA as template. As controls, a parallel set of reactions was run without reverse transcriptase and one reaction did not contain RNA template. A SuperScript first-strand synthesis system (Invitrogen) was used for cDNA synthesis. Power SYBR Green PCR master mix (Applied Biosystems) was used for qPCR which was performed on an ABI 7500 Fast PCR system (software version 2.3). The run conditions were 95°C for 15 s, 58°C for 15 s, and 60°C for 60 s for each cycle. Expression of TXNRD1 mRNA in each sample was calculated relative to TXNRD1 expression in untreated cells. Values for TXNRD1 expression were normalized against expression values for the GAPDH housekeeping gene. The Pfaffl equation was used for calculation of relative expression.
RESULTS
Readthrough of the HIV-1 nef 3’-UGA stop codon in HEK 293T cells transfected with nef-ATI-GFP constructs
GFP production from two versions of a wild-type HIV-1 nef expression construct designed to read past the conserved UGA stop codon into a downstream GFP domain [a and b in Figure 3] was compared to that from several controls: A negative control in which an in-frame TAA stop codon was inserted between the nef and GFP domains [c in Figure 3], the parent GFP expression plasmid as a positive control (d) and a second negative control consisting of untransfected cells (e). The relative GFP production calculated using ImageJ is shown in panel f. Both the ATI1 and ATI1+2 constructs showed highly significant levels of stop codon readthrough relative to the negative control ATIstop construct, which was identical to the ATI2 construct except for two in-frame stop codons designed to prevent further translation into the GFP coding region [Figure 2]. Differences between results for pNefATI1, pNefATI2, and the parent GFP construct (as 100% readthrough control) were all statistically significant at P < 0.001. Unexpectedly, the inclusion of the second smaller anti-TXNRD1 region ATI2 in the construct leads to a decrease rather than the expected enhancement of readthrough (see Discussion). As expected, there was undetectable GFP expression from the untransfected control cells (E).

- Readthrough of the HIV-1 nef 3’-UGA codon. The photomicrograph panels (a-e) show GFP fluorescence in HEK 293T cells, transfected with three different ATI plasmid vector constructs and controls. (a) pNefATI1; (b) pNefATI2; (c) pNefATIstop; (d) Parent GFP construct; (e) untransfected cells; (f) Bar graph showing the GFP intensity of each transfection condition calculated using NIH ImageJ Software.
Selenium dependence of stop codon readthrough
Figure 4 shows the effect of varying the pretreatment selenium concentration on GFP expression from the ATI1 construct, which is a measure of the efficiency of UGA readthrough. HEK 293T cells were pretreated with different concentrations of exogenous selenium as sodium selenite, ranging from 0 to 80 nM final concentration, before transfection with the ATI1 plasmid. The results from Figure 3 show that for pNefATI1, even without added selenite, GFP expression in standard culture medium is significantly higher than the untreated control (P < 0.0001). With additional selenium supplementation, as shown in Figure 4, the amount of readthrough product more than doubles going from 0 to 20 nM selenium, for which the difference is also significant (a 119% increase, P < 0.0001). However, GFP production levels for 20, 50, and 80 nM selenium are not significantly different from each other. Thus, at 20 nM of added exogenous selenium, the mechanism appears to be saturated, as no further increase is observed at higher concentrations.

- Added selenium enhances stop codon readthrough from the ATI-1 plasmid construct. Results show that supplementation of the basal media with additional selenium as sodium selenite has a significant effect on the stop codon readthrough (P < 0.0001), resulting in higher levels of GFP expression. However, the maximal readthrough was observed at the lowest level of selenium (20 nM) and the readthrough did not change significantly at higher selenium concentrations. Even at the 20 nM concentration, addition of sodium selenite essentially doubles UGA stop codon readthrough relative to unmodified cell culture medium, as measured by GFP production.
Flow cytometry analysis of GFP expression from nef-ATIGFP constructs versus controls
Flow cytometry was used to obtain a more accurate quantitative assessment of the nef UGA codon readthrough efficiency in the nef-ATI1 construct, relative to the parent GFP expression construct. As shown in Figure 5, the P1 population of cells transfected with pNefATI1 had a mean GFP fluorescence of 11,443 ± 1242, as compared to the background of 1304 ± 553 in untransfected control cells, and 55,076 ± 11,111 in the cells transfected with the parent GFP construct (a standard for 100% readthrough efficiency as it lacks any in-frame UGA codon). This difference is highly significant (P < 0.0001) and suggests a readthrough efficiency on the order of (11,443–1304)/(55,076– 1304) × 100 = 18.9% for the wild type HIV-1 nef 3’-UGA.

- Flow cytometry analysis of HIV-1 nef stop codon readthrough. A (Top): HEK 293T Cells transfected with pNefATI1 vector. FITC-A represents green fluorescence (GFP). The P1 population had a mean FITC-A of 11,443 ± 1,242. B (Middle): Cells transfected with EGFP-N3 plasmid. The P1 population had a mean FITC-A of 55,076 ± 11,111. C (Bottom): Untransfected cells (background). The mean FITC-A of P1 was 1,304 ± 553. Stop codon readthrough efficiency was thus 18.9% (see text).
siRNA knockdown of TXNRD1 mRNA decreases nef 3’-UGA codon readthrough
To assess the hypothesized role of TXNRD1 mRNA in facilitating the UGA readthrough demonstrated in Figures 3-5, anti-TXNRD1 siRNA was used to knock down TXNRD1 mRNA levels, which we predict should lead to decreased GFP production from the nef ATI1 construct.
Figure 6 shows the results of a preliminary experiment used to determine the optimal transfection reagent and concentrations for the subsequent siRNA studies. This resulted in the choice of the siPort Amine at 0.6 μl per well as the transfection reagent for the siRNA TXNRD1 knockdown experiment as shown in Figure 7.

- Selection of transfection reagent and optimization of the transfection reagent volume using HEK 293T cells transfected with pNefATI1 EGFP-N3 vector. EVOS GFP fluorescence images (a-f) are cells transfected with 0.3 ul amine, 0.6 ul amine, 0.15 ul amine, 0.5 ul NeoFX, 1.2 ul NeoFX, and 0.15 ul NeoFx, respectively. Image g represents untransfected cells.

- Effect of siRNAs on GFP production from the pNefATI1 construct. Panels A-E show EVOS microscopy images of each transfection condition: (a) pNefATI1 with no siRNA. (b) With added negative control scrambled siRNA. (c) With positive control anti-GAPDH siRNA. (d) With anti-TXNRD1 (TR1) siRNA. (e) Untransfected cells. (f) Bar graph showing average GFP expression from ten images of each treatment calculated using NIH ImageJ software.
In Figure 7, the effects of three different siRNAs on GFP production from the pNefATI1 plasmid are shown, as compared to pNefATI1 with no siRNA, and untransfected controls. The three siRNAs used were anti-TXNRD1, a random “scrambled” siRNA, and anti-GAPDH as a housekeeping gene control. The results show that the anti-TXNRD1 siRNA produces a significantly greater decrease in pNefATI1-associated GFP production than either the scrambled or anti-GAPDH siRNAs. Compared to panel A (GFP production from pNefATI1 with no added siRNA), we observe a slight but non-significant decrease in GFP production in the presence of scrambled siRNA (panel B), a greater (~50%), significant decrease with the positive control anti-GAPDH siRNA (Panel C; P < 0.001), but the largest decrease of ~67% was with the anti-TXNRD1 siRNA (Panel D; P < 0.0001). There is no detectable GFP production from the untransfected control cells (Panel E). The average GFP values from the different conditions are compared in the bar graph in Panel F, from which the stated p values and percent decreases were calculated.
Validating TXNRD1 mRNA knockdown using RT-PCR
To validate the siRNA knockdown of TXNRD1 in the previous experiment [Figure 7], TXNRD1 expression in cells treated with the anti-TXNRD1 siRNA was assessed using qPCR, as compared to TXNRD1 expression in untreated cells, and cells treated with the scrambled siRNA, and cells transfected with the pNefATI1 construct alone. As shown in Figure 8, the presence of anti-TXNRD1 siRNA lead to the largest and most significant decrease in TXNRD1 mRNA levels in treated cells, a decrease of about 28% under the conditions used (P < 0.0001). In cells treated with either the scrambled siRNA or pNefATI1 there was a small (~9%) and less significant decrease (P < 0.01) in TXNRD1 mRNA.
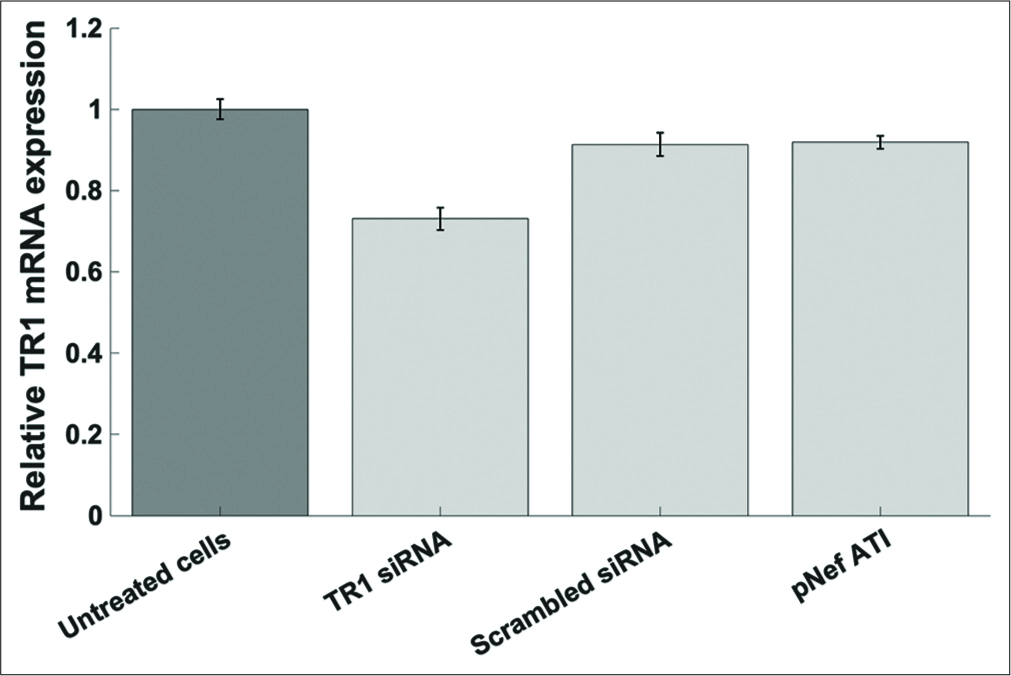
- TXNRD1 mRNA expression assessed by qPCR. Compared to untreated cells, an approximately 27% knockdown of TXNRD1 mRNA relative to untreated cells (P < 0.0001) was observed in the sample treated with anti-TXNRD1 siRNA (TR1 si/RNA). This confirms the knockdown of TXNRD1 mRNA that may contribute to the decrease in nef 3’-UGA readthrough in the presence of anti-TXNRD1 siRNA [Figure 7].
DISCUSSION
The results of the current study may be summarized as follows:
Using full length expression constructs of the HIV-1 nef gene containing downstream sequence elements extending as much as 130 nucleotides past the terminal 3’-UGA stop codon, cloned upstream of GFP (as a reporter for translational readthrough), we have demonstrated that this UGA codon is suppressed at detectable efficiency [Figures 3-5], estimated by FACS analysis to be about 19% by comparison to GFP production from the parent GFP construct [Figure 5]
The nef expression constructs in question, pATI-1 and pATI-2, contain sequences of antisense complementarity to human TXNRD1, identified previously as regions of potential ATI targeted by HIV-1, as shown schematically in Figure 1. The dependence of nef 3’-UGA readthrough on TXNRD1 mRNA is supported by our finding that, in the presence of anti-TXNRD1 siRNA, there is a substantial and significant decrease (~67%, P < 0.0001) in UGA readthrough from the nef pATI-1 construct; this decrease is also significantly larger than the effect of either of the control siRNAs [Figure 6]. This result supports our hypothesis that antisense tethering of TXNRD1 mRNA may contribute to the translational read through
When the selenium concentration of the culture media was increased above basal levels by the addition of 20nM or greater sodium selenite, nef 3’-UGA readthrough increased by over 100%, supporting a role for selenium in the readthrough mechanism. However, our results do not address the question of whether this is due to the incorporation of Sec at the UGA codon, as hypothesized previously,[1,4] or by some other mechanism.
These results strongly support our earlier predictions of translational readthrough of the HIV-1 nef 3’-stop codon, and a potential role for selenium.[1] However, they fall short of demonstrating that at least a subpopulation of translational readthrough products may contain Sec at the location of the UGA codon. UGA is known to be a very “leaky” stop codon, and various amino acids with related codons (e.g. Trp, Cys, and Ser) have been observed at this codon position in known examples, including mixtures of the various possible products, for example, as reported by Chittum et al. for beta globin.[12]
This key finding is also consistent with experimental evidence for an extended isoform of nef obtained by researchers at the University of Rochester, NY, in which the nef extension was identified by immunohistochemical methods, in both post-mortem HIV+ brain slices and in vitro translated nef constructs from actual patient isolates (Dr. Benjamin Blumberg, personal communication).
The evidence presented here obtained using siRNA in regard to a possible role for TXNRD1 mRNA in the observed readthrough is positive, since there is a significant ~67% decrease (P < 0.0001) in GFP production from the pNefATI1 construct in the presence of anti-TXNRD1 siRNA [Figure 7], which is significantly greater than the reduction seen with either the anti-GAPDH control or the scrambled siRNA negative control; the decrease seen with the scrambled siRNA is not significantly different from no siRNA at all. The effect of the anti-GAPDH siRNA may simply reflect a decrease in overall cell activity produced by the targeting of an essential housekeeping gene.
We verified TXNRD1 knockdown by this commercial anti-TXNRD1 siRNA using RT-PCR [Figure 8], which in our hands showed only a 27% decrease in TXNRD1 mRNA levels. The manufacturer claims that the knockdown achieved with this siRNA should be greater, about 70%, which would be more consistent with the potency we observed for its effect on inhibition of GFP production from the pNefATI1 plasmid (a 67% decrease); however, it is possible that only a 27% decrease in TXNRD1 mRNA could have a somewhat amplified effect on a downstream system.
One important observation from Figure 8 is that pNefATI1 produced only a minimal (9%) knockdown of TXNRD1 mRNA, similar to that produced by the scrambled siRNA. This is despite the fact that pNefATI1 does express an mRNA with a region of antisense complementarity to TXNRD1 mRNA. However, antisense and miRNA type interactions do not necessarily lead to degradation of a target mRNA, which depends on a high degree of complementarity of the 2 RNAs; when the antisense interaction is not perfect, it can be the basis of ribosomal interference with the target protein synthesis, without mRNA degradation.[13] Thus, our results suggest that the antisense targeting of TXNRD1 mRNA by the HIV-1 nef 3’-region, in addition to serving a possible tethering function for TXNRD1 SECIS capture[1], may also lead to inhibition of TXNRD1 protein synthesis in infected cells, but not through degradation-mediated downregulation of TXNRD1 mRNA.
One unexpected result was that the readthrough efficiency was LESS when both anti-TXNRD1 ATI regions were present in the nef construct, as opposed to just ATI1 alone [Figure 4]. Because in nef ATI1 the 3’-UGA is right near the center of an ~40 residue sequence that is complementary to a region of TXNRD1 mRNA, during translation up to and past the stop codon, which we have now shown does occur [Figures 3-5], ATI1 would have to become unwound as that region of the nef mRNA enters the ribosome. Thus, we suspected that the second downstream ATI region, though smaller (only 13 base pairs in length), might be important to keep the TXNRD1 mRNA and its SECIS element in proximity to the ribosome. However, it is also possible that the dynamics of the engagement of the TXNRD1 SECIS to the ribosome are such that they could continue to function in the recoding of the heterologous viral UGA, for at least a short time after the unwinding of ATI1. Hence, ATI2 may not be particularly important. However, it is also possible that the reason for the decrease in readthrough efficiency of the construct that includes ATI2 is simply due to the fact that, in order to engineer this construct to include the ATI2 region, while at the same time permitting readthrough into the downstream GFP region, a region of viral 3’-RNA that normally has non-coding roles must be translated. Thus, codon usage in this region may be non-optimal, affecting translation efficiency in our ATI2 construct. In contrast, readthrough of the 3’-UGA, which we have now observed in transfected cells, would by necessity lead to C-terminal extension of the nef coding region by up to 33 amino acids, so that extended nef codon usage in the entire region spanning ATI1 has presumably been optimized for efficient translation during viral evolution. That is not the case for the region spanning ATI2, due to the TAA stop codon at its 5’ end [Figures 1 and 2]. In addition, the noncoding far 3’-end of the HIV-1 mRNA contains a number of RNA hairpin structures with diverse regulatory functions, the unwinding of which might slow translation around the ATI2 region.
An alternative explanation for GFP expression from these constructs could involve initiation of GFP protein translation at an internal start codon. Although there is a candidate methionine in the N-terminal region of the GFP coding sequence, there are no in-frame Met codons past the 3’-UGA of nef in any of the included viral sequences. Hence, the possibility of internal initiation of GFP synthesis fails to explain our observations, because if it was happening from the GFP internal Met, GFP production would also be observed with the pNefATIstop control construct, which is not the case [Figure 3c].
Finally, it must be emphasized that the current results showing a link between selenium biology and the expression of an isoform of a gene (nef) that is central to HIV-1 pathogenesis should be interpreted in the light of an extensive body of evidence linking HIV disease progression and outcome to selenium status. Observations of selenium abnormalities in AIDS patients date back almost as far as the discovery of the virus in the 1980s (e.g.,[14,15]). Later studies have linked HIV-associated mortality risk to serum selenium levels,[16-18] and demonstrated various clinical benefits for selenium supplementation, either alone or as part of a micronutrient formulation.[19-21]
Nor is HIV-1 an exceptional case in this regard. A number of predominantly RNA viruses have been shown in animals or humans to have either increased frequency and/or severity of cases in low selenium areas or individuals, or some measurable clinical or survival benefit from selenium supplementation (as reviewed,[22-24]). The most recent example is seen with the COVID-19 pandemic, for which Zhang et al. have now demonstrated a highly significant association between the outcome of SARS-CoV-2 infection and previously documented regional selenium status in Chinese cities.[25]
Based on our results to date, and as proposed previously,[26] we predict that antisense interactions with TXNRD isoforms will be identified for a number of other RNA viruses, and that this may prove to be a general mechanism for RNA viruses to enhance RNA levels for virus production, by partially inhibiting the conversion of ribonucleotides to 2- deoxyribonucleotides. Because ribonucleotide reductase uses the thioredoxin system as a hydrogen donor, antisense knockdown of TXNRD isoforms could tip the balance in favor of RNA production, by inhibiting the regeneration of reduced thioredoxin. This biochemical role of selenium in mammals may prove to be a significant factor contributing to the increased virulence of some RNA viruses in selenium-deficient hosts.
CONCLUSION
We have provided compelling evidence that sequences within the HIV-1 nef mRNA, with its proximal downstream elements, are sufficient to permit readthrough suppression and thereby extension of the nef protein into the downstream region. The only non-native element introduced into the pNefATI1 construct was mutation of the TAA stop codon at the end of that extension region, to permit translation of the downstream GFP reporter gene. We have also demonstrated that addition of a low concentration (20 nM) of selenium as sodium selenite can increase the readthrough efficiency by over 100%, and that knockdown of TXNRD1 mRNA can reduce the efficiency of the stop codon readthrough by about two-thirds. These findings are consistent with a role for TXNRD1 mRNA and selenium-based cellular mechanisms in the readthrough event, but further studies will be required before we can say with certainty that virus/host mRNA tethering reactions are involved in these observations – however, it has been strengthened as a hypothesis.
Data availability statement
The calculated relative fluorescence values and FACS data used to support the findings of this study are included within the article.
Declaration of patient consent
Patient’s consent not required as there are no patients in this study.
Financial support and sponsorship
Dr. Arthur and Bonnie Ennis Foundation, Decatur, IL.
Conflicts of interest
There are no conflict of interest.
References
- Cellular selenoprotein mRNA tethering via antisense interactions with Ebola and HIV-1 mRNAs may impact host selenium biochemistry. Curr Top Med Chem. 2016;16:1530-5.
- [CrossRef] [PubMed] [Google Scholar]
- Structure and mechanism of mammalian thioredoxin reductase: The active site is a redox-active selenolthiol/selenenylsulfide formed from the conserved cysteine-selenocysteine sequence. Proc Natl Acad Sci U S A. 2000;97:5854-9.
- [CrossRef] [PubMed] [Google Scholar]
- The molecular biology of selenocysteine. Biomol Concepts. 2013;4:349-65.
- [CrossRef] [PubMed] [Google Scholar]
- Nutrition, HIV, and drug abuse: The molecular basis of a unique role for selenium. J Acquir Immune Defic Syndr. 2000;25(Suppl 1):S53-61.
- [CrossRef] [Google Scholar]
- Evolutionary history of selenocysteine incorporation from the perspective of SECIS binding proteins. BMC Evol Biol. 2009;9:229.
- [CrossRef] [PubMed] [Google Scholar]
- Bipartite signal for read-through suppression in murine leukemia virus mRNA: An eight-nucleotide purine-rich sequence immediately downstream of the gag termination codon followed by an RNA pseudoknot. J Virol. 1992;66:5127-32.
- [CrossRef] [PubMed] [Google Scholar]
- Molecular modeling and in vitro activity of an HIV-1-encoded glutathione peroxidase. Proc Natl Acad Sci U S A. 2000;97:6356-61.
- [CrossRef] [PubMed] [Google Scholar]
- An HIV-1 encoded peptide mimics the DNA binding loop of NF-kappaB and binds thioredoxin with high affinity. Mutat Res. 2005;579:133-48.
- [CrossRef] [PubMed] [Google Scholar]
- Anti-apoptotic activity of the glutathione peroxidase homologue encoded by HIV-1. Apoptosis. 2004;9:181-92.
- [CrossRef] [PubMed] [Google Scholar]
- Functional characterization of the eukaryotic SECIS elements which direct selenocysteine insertion at UGA codons. EMBO J. 1993;12:3315-22.
- [CrossRef] [PubMed] [Google Scholar]
- Rabbit beta-globin is extended beyond its UGA stop codon by multiple suppressions and translational reading gaps. Biochemistry. 1998;37:10866-70.
- [CrossRef] [PubMed] [Google Scholar]
- MicroRNAs and small interfering RNAs can inhibit mRNA expression by similar mechanisms. Proc Natl Acad Sci U S A. 2003;100:9779-84.
- [CrossRef] [PubMed] [Google Scholar]
- Selenium deficiency in the acquired immunodeficiency syndrome. J Parent Enteral Nutr. 1986;10:405-7.
- [CrossRef] [PubMed] [Google Scholar]
- Abnormalities of blood selenium and glutathione peroxidase activity in patients with acquired immunodeficiency syndrome and aids-related complex. Biol Trace Elem Res. 1988;15:167-77.
- [CrossRef] [PubMed] [Google Scholar]
- High risk of HIV-related mortality is associated with selenium deficiency. J Acquir Immune Defic Syndr Hum Retrovirol. 1997;15:370-4.
- [CrossRef] [PubMed] [Google Scholar]
- Mortality risk in selenium-deficient HIV-positive children. J Acquir Immune Defic Syndr Hum Retrovirol. 1999;20:508-13.
- [CrossRef] [PubMed] [Google Scholar]
- Serum selenium predicts outcome in HIV infection. J Acquir Immune Defic Syndr Hum Retrovirol. 1995;10:392.
- [CrossRef] [PubMed] [Google Scholar]
- A randomized trial of the impact of multiple micronutrient supplementation on mortality among HIV-infected individuals living in Bangkok. AIDS. 2003;17:2461-9.
- [CrossRef] [PubMed] [Google Scholar]
- Effect of micronutrient supplementation on disease progression in asymptomatic, antiretroviral-naive, HIV-infected adults in Botswana: A randomized clinical trial. JAMA. 2013;310:2154-63.
- [CrossRef] [PubMed] [Google Scholar]
- Effect of selenium supplementation on CD4+ T-cell recovery, viral suppression and morbidity of HIV-infected patients in Rwanda: A randomized controlled trial. AIDS. 2015;29:1045-52.
- [CrossRef] [PubMed] [Google Scholar]
- Selenium, selenoproteins and viral infection. Nutrients. 2019;11:2101.
- [CrossRef] [PubMed] [Google Scholar]
- Selenium and RNA virus interactions: Potential implications for SARS-Cov-2 infection (Covid-19) Front. Nutr. 2020;7:164.
- [CrossRef] [PubMed] [Google Scholar]
- Dietary selenium in adjuvant therapy of viral and bacterial infections. Adv Nutr. 2015;6:73-82.
- [CrossRef] [PubMed] [Google Scholar]
- Association between regional selenium status and reported outcome of COVID-19 cases in China. Am J Clin Nutr. 2020;111:1297-9.
- [CrossRef] [PubMed] [Google Scholar]
- RNA viruses vs. DNA synthesis: a general viral strategy that may contribute to the protective antiviral effects of selenium. In: Preprints. 2020.
- [CrossRef] [Google Scholar]




